A dimensionality reduction technique particularly well suited for visualizing data. (For references, see https://lvdmaaten.github.io/tsne)
The parameters that were used for running t-SNE here are: 50 initial dimensions, perplexity of 30, and theta of 0.5. For datasets with <= 5000 samples, the standard t-SNE algorithm is used. For larger datasets, the Barnes-Hut algorithm is employed.
A dimensionality reduction technique in which the two principal components are chosen to have the largest possible variance.
To analyze relationships between perturbations, we utilize the framework of connectivity. A connectivity score between two perturbations quantifies the similarity of the cellular responses evoked by these perturbations. A score of 1 means that these two perturbations are more similar to each other than 100% of other perturbation pairs. A score of -1 means that these two perturbations are more dissimilar to each other than 100% of other perturbation pairs.
See a heatmap of connections between individual perturbagens in cell lines and all other perturbagens used for the P100 assay or the GCP assay. The tutorial describes the features of the heatmap.
Bring data, in GCT format, from your own P100 or GCP studies to query against our datasets.
Introspect means querying your dataset against itself. Make sure to "Include Introspect" if you would like to see connections within your dataset (in addition to connections between your dataset and Touchstone-P).
In computing connectivity, biological or technical replicates can be aggregated together. Please select which metadata fields should be used to recognize replicates. For example, if you wish to distinguish between different doses of the same compound, make sure to select "pert_dose" (or something similar) as one of the metadata fields by which to group replicates. The possible metadata fields by which to group replicates only appear after you have upload your GCT and selected "Yes" for "Are there replicates in your data?".
Matched mode: When running GUTC, incorporates cell-line information to match
query data against matching cell types in Touchstone. Currently
this includes the following 9 cell types : [A375, A549,
HEPG2, HCC515, HA1E, HT29, MCF7, PC3, VCAP].
Unmatched mode (recommended): When running GUTC, does not incorporate cell-line information when querying
the data against Touchstone signatures.
L-Build ("Light" Build): All levels of L1000 data up to aggregated signatures.
Full Build: All levels of L1000 data up to aggregated signatures, as well as all relevant additional analyses
of the data (Introspect, t-SNE, PCA, etc.).
When querying Touchstone, Feature Space determines what set of genes to query against. When perturbagens are profiled on the L1000 platform, Landmark is recommended. When the queries you wish to use are not landmarks, use BING instead.
Root location within a brew folder that contains the instance matrices and the brew_group folder. Default is brew/pc
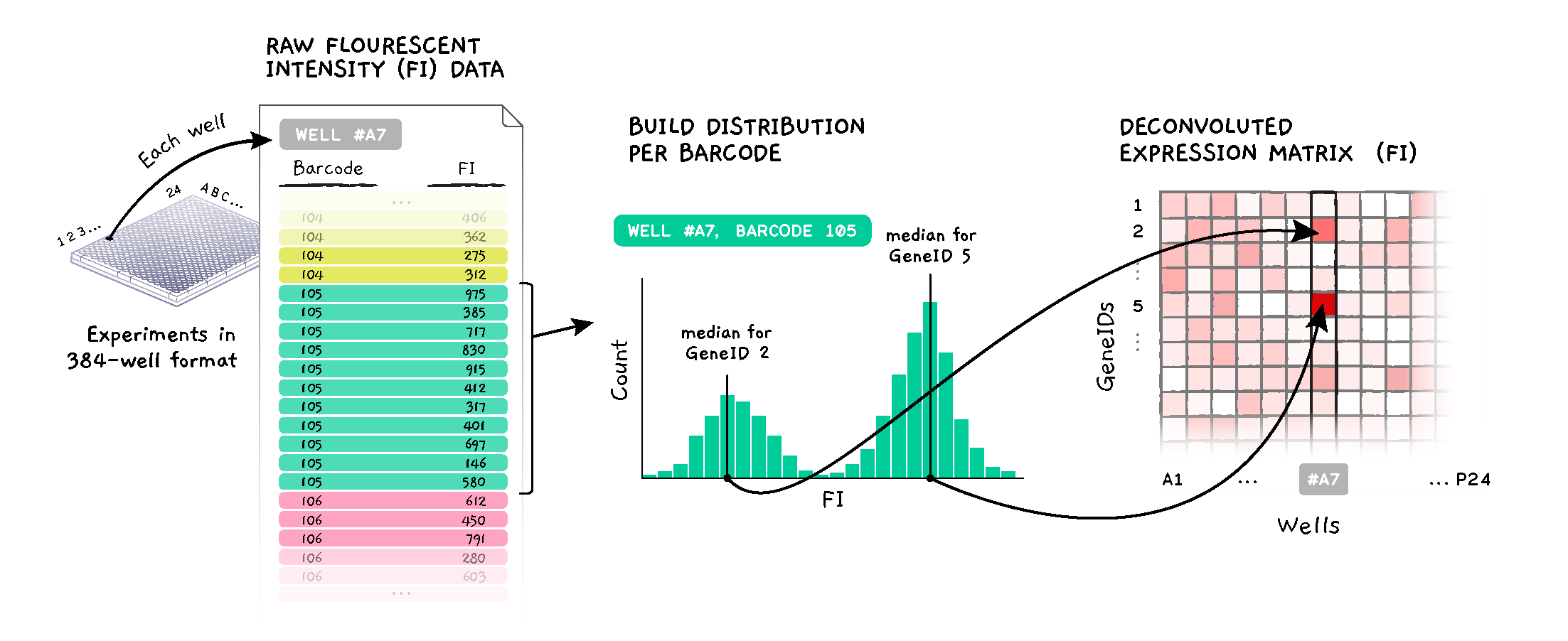
List of expected treatment doses in micromolar as a listmaker list. If provided, dose discretization is applied to the pert_dose metadata field to generate a canonicalized pert_idose field. Note this assumes that the pert_dose annotations are in micromolar.
Generates TAS plots and connectivity heatmap of preliminary callibration plates to identify the most suitable experimental conditions of specified parameters. Tool should be run on small pilot experiments, with a variety of experimental parameters such as seeding density and time point. Plots can also be decoupled by parameters such as cell id.
Column filter to sig_build_tool as a listmaker collection
The name of the build used when generating all associated files and folders (e.g. <BUILD_CODE>_metadata). For this reason, the code must be filename compatible.
When merging replicates for L1000, several versions of the merged data are made. This parameter determines which version to use when creating your build. by_rna_well is the default. by_rna_well is recommended.
All data is from the Cancer Cell Line Encyclopedia resource. Expression data was released 15-Aug-2017, copy number data is dated 27-May-2014, and mutational data is dated 15-Aug-2017.
Feature Mapping: Ensembl Ids from the source data were mapped to Entrez Gene Ids using gene annotations from NCBI (downloaded on 02-Mar-2016).
Normalization: RNAseq RPKM values were log2 transformed using log2(max(RPKM, eps)). The data were then normalized
such that the expression values were comparable across cell lines, by minimizing technical variation
and equalizing their distributions (for details of the normalization, see LISS and QNORM entries in the Connectopedia glossary). Post-normalization, the expression values range between 4 and 15 log2 units, with 4 indicating that
a gene is minimally or not expressed and 15 indicating the maximum readout.
Z-scores: The number of standard deviations that a gene is above or below the population mean is called its z-score. The "robust" z-score is resistant to outliers by using median instead of mean and median absolute deviation (MAD) instead of standard deviation. The reference population used to compute the median and MAD for a
particular gene is all CCLE lines with data for that gene.
Z-scores Within Primary Site: Similar to z-scores,
but the reference population used to compute the median and MAD is all CCLE lines from the same lineage with data for that gene.
All scores indicated are in log 2 ratios to reference, binned using the heuristics described in CNVkit.
Deletion: score < -1.1
Loss: -1.1 ≤ score ≤ -0.25
No change: -0.25 < score < +0.2
Gain: +0.2 ≤ score < +0.7
Amplification: +0.7 ≤ score
Access a suite of analysis apps by clicking on the menu (or type command-K to open)
Explore the Connectivity Map by typing here and pressing Enter (see instructions below the search box)
Switch between running a single query and running a batch query.
Give each query a descriptive name that will help you identify your results.
Tip: Each list can have a different number of genes; in fact, you can run a query with only one list (up OR down).
Your query will take about 5 minutes to process; check the History section in the Menu for your results!
Valid genes used in the query have HUGO symbols or Entrez IDs and are well-inferred or directly measured by L1000 (member of the BING gene set). Valid genes not used in a query are those that have a valid HUGO or Entrez identifier but are not part of the BING set. Invalid genes do not have HUGO or Entrez IDs.
Give each query a descriptive name that will help you identify your results.
Your query will take about 5 minutes to process; check the History section in the Menu for your results!
The sig_fastgutc_tool is a reimplementation of our query algorithm that enables faster query results, especially at larger batch sizes. It is the result of crowd-sourced contest. It is currently in beta mode.
Filter datasets by category to see only those of interest.
Data Icons identify published and proprietary datasets.
Click on a row to see a summary of that dataset, including cell lines and treatment conditions, assay type, and dates.
Arrange the table to display the information most important for your work, and add key datasets to favorites.
View details about the collection as a whole and about individual compounds.
View subsets of compounds based on mechanism, drug target, or known disease application.
Purity is assessed by ultra-performance liquid chromatography-mass spectrometry (UPLC-MS) of compounds after receipt from the vendor.
Status as of publication of this resource (March 2017). We will be updating this but let us know if you notice a discrepancy.
Click on a compound to see details about its structure, mechanism, targets, approval status, and vendor.
Mouse over this graphic to see the classes of proteins targeted by drugs in the hub.
Search CMap for connections. First, search for a perturbagen and check the box to select it. Then view its connections as a list or heatmap.
This is the current count of perturbagens in the reference (touchstone) dataset.
Select data from perturbagens grouped by their MoA or role in the cell.
Choose a perturbagen type, or view them all.
Touchstone is our reference dataset, made from well-annotated perturbagens profiled in a core set of 9 cell lines.
Detailed List is unavailable for Touchstone v1.1.1.1. A new data visualization approach is in development, but to get results in a table format (similar to Detailed View), please click on Heat Map and download the dataset as a GCT file that can be viewed in Excel or similar apps. Please see here for a detailed explanation.
Use the Command app to retrieve the most up-to-date CMap information. Type a slash (/) to see the list of commands available. Select a command, and type the MoA, gene, perturbagen, or other keyword after it to specify your request.
Articles are tagged with topics. Click on a topic tag to see all related articles.
Look it up! A quick reference guide of CMap terms and their meanings.
Email us with your questions.
Click on the heading to read all the articles in this section on a single page, or open each article separately.
Click on a heading to open a menu of articles.
Articles with the same tag(s) as this article can be opened here. To see all articles associated with any tag, click on the tag from the list below.
Each article is tagged with key words that describe its content.
Underlined words link to their definition in the CMap glossary.
Your feedback helps us make Connectopedia more useful.
Average transcriptional impact
TAS is a metric that incorporates the signature strength (the number of significantly differentially expressed transcripts) and signature concordance (the reproducibility of those changes across biological replicates) to capture activity of a compound. The score is computed as the geometric mean of the signature strength and the 75th quantile of pairwise replicate correlations for a given signature. Prior to computing the geometric mean, the signature strength is multiplied by the square root of the number of replicates. This serves to mitigate score shrinkage with increasing replicate number and allows TAS values derived from signatures of different numbers of replicates to be compared with each other.
Signature diversity
Thick black bars signify Transcriptional Activity Scores greater than or equal to 0.5; thinner black bars denote scores less than 0.5. Absence of a bar means no data available. Colored lines (chords) signify similar connectivity scores between cell lines; red for positive connectivity scores of 80-100 (pale to intense color according to the score); blue for negative connectivity. Chords are only shown when TAS scores are > 0.5; thus absence of a chord either means that the perturbagen TAS score is very low, or that no data is available. Chords for individual cell lines can be isolated from the rest of the figure by hovering over the cell line name.
Baseline expression of this gene in each cell line is represented as a z-score (top numbers). Scores were calculated using robust z-score formula:
z-scorei = ( xi - median( X ) )/( MAD( X ) * 1.4826 ),
where:
xi is expression value of a given gene in i-th cell line
X = [ x1, x2 ... xn ] is a vector of expression values for a given gene across n cell lines
MAD( X ) is a median absolute deviation of X
1.4826 is a constant to rescale the score as if the standard deviation of X instead of MAD was used
Median and MAD expression values were calculated using RNA-Seq profiles from a total of 1022 cell lines, comprising data from the Cancer Cell Line Encyclopedia (CCLE; Barretina, et al.) and cell lines nominated by the CMap team. Plots show z-score values only for the core LINCS lines used by CMap in L1000 experiments. Light red or light blue regions indicate positive or negative outlier expression, respectively, of the gene relative to the other lines shown; z-score of a positive outlier in the corresponding cell line is in dark red and a negative outlier is in dark blue.
Summary class connectivity shows a boxplot that summarizes the connectivity of a class. Each data point, shown as a light gray dot, represents the median value of connectivity of one member to the other class members. (This corresponds to the median for each row, excluding the main diagonal, in the heatmap shown below.) The box is the distribution of those data points, where the box boundary represents the interquartile range, the vertical line within the box is the median, and the whiskers reflect the minimum and maximum values of the data (exclusive of extreme outliers, which may appear beyond the whiskers).
Connectivity between members of class is a standard heat map of the connectivity scores, summarized across cell lines, between members of the class, where dark red represents the highest positive scores and deep blue the highest negative scores. Individual scores are revealed to the left below the map by hovering over each cell of the map.
Class inter-cell line connectivity is a plot of the median (black line) and Q25-Q75 connectivity scores (blue area around black line) for each cell line as well as the summary scores across cell lines. In some cases perturbations have not been tested in every cell line; the absence of data is indicated by a “0” for that cell line. The example shown reveals that these estrogen agonists show the strongest connectivity to each other in MCF7, a human breast cancer cell line that expresses the estrogen receptor.
Profile status
Colored portion of top bar indicates the Broad assays in which this compound has been profiled.
L1000 cell/dose coverage
For compounds profiled by L1000, cell lines and dose range for which signatures are available are indicated by dark gray bars (lighter gray bar indicates no data is available for that cell line/dose combination). A bar displayed one row above the 10 uM row indicates that doses higher than 10uM were tested. The 6 rows correspond to 6 canonical doses: 20 nM, 100 nM, 500 nM, 1 uM, 2.5 uM, and 10 uM. (In some cases non-canonical doses were tested; these are rounded to the nearest canonical dose for the purpose of this display. For example, if the dose tested was 3.33uM, the 2.5uM bar is shown in dark gray here.)

Log out
Log in
Forgot password?
To reset your password, enter your email address and click Submit.
Sign up
Check your email
Your account has been activated. Please log in.
Your password has been reset!
Settings
Privacy Policy
Terms and Conditions
We've sent an email to bob@acme.com, from clue@broadinstitute.org.
Click the link in the email to reset your password.
Go to CMap homepage.
Tools
Projects
My Collaborations
Public Projects
Developer
For Developers
Help
Helpful Links
Menu
|
Announcements
See all